

Taxonomic
classification with CREST
Bioinformatics for metagenomic
analyses and environmental sequencing
Oslo, 18-22 March 2013
Anders Lanzén
Overview
1. Familiarise yourself
with CREST installation
2. Download the data
3. De-noising the
pyrosequencing raw data using AmpliconNoise (optional)
4. Aligning files to the
reference database (SilvaMod) using Megablast
5. Taxonomic
classification using LCAClassifier
6. Comparison of
composition between datasets
7. Extracting sequences
from a taxon
8. Classification using
Greengenes
9. Classification with
MEGAN (optional)
1. Familiarise yourself with CREST installation
LCAClassifier is the program in the package CREST (Classification Resources for Environmental Sequence Tags) that is responsible for taxonomical classification of sequences based on Megablast alignments to a reference database. On the course lab computers, this program is installed in the directory /home/qiime/Desktop/LCAClassifier. The installation directory contains a configuration file that defines the location of database files that the LCAClassifier needs in order to map Megablast results to a taxonomy. Find and open the file lcaclassifier.conf in the LCAClassifier subdirectory parts/etc, either using the graphical or command line interface.
As you can see this configuration file contains references to two different reference databases / taxonomies. By default, two databases are available: the modified version of the Silva Taxonomy, called SilvaMod and Greengenes. These are downloaded when LCAClassifier is installed under the folder parts/flatdb. Each database contains the following files, named like the database itself, e.g. for SilvaMod:
1)
silvamod.fasta.n* (.nhr, .nin and
.nsq) –binary search index files for BLAST /
Megablast. The FASTA-formatted sequences themselves are not needed by BLAST and
not downloaded by default, to speed up the installation.
2)
silvamod.tre - a text file in Newick tree format that defines the topology of
the taxonomical tree
3)
silvamod.map - a tab-separated text file that specifies a name and rank for each
taxon ID in the tree-file
Now, use the command line shell (Terminal
window) create a directory for this tutorial under your home directory, called
e.g. CREST_Tutorial:
mkdir Desktop/CREST_Tutorial
cd Desktop/CREST_Tutorial
2. Download the data
In this tutorial we will classify two sets of amplicon sequence data. These sequence sets are called LC_Prefilter and LC_Final and originate from an alkaline soda lake in Ethiopia (“LC”=Lake Chitu). The difference between the two is that the DNA for the first is collected from a more rough filter and the second results from this filtrate, collected using a more fine filter (so, not a noise-filter but an actual, real filter for water samples). Thus, we expect to only catch planktonic microorganisms in the second dataset.
First, however, we need to download the data using wget.
wget
http://services.cbu.uib.no/supplementary/crest/SodaLakeAmplicons.tar.gz
Then uncompressed the three files:
tar xvzf SodaLakeAmplicons.tar.gz
As you can see, the folder contains three files: LC_Prefilter_F_Good.fa, LC_Final_F_Good.fa and LC.sff. The first two are FASTA-files, de-noised and annotated using AmpliconNoise to represent only unique sequences with different numbers of reads. The second is in SFF-format (standard flowgram format), which is the raw data from 454 pyrosequencing. If you want, and if the service is fast enough, you can submit this SFF-file to the AmpliconNoise web server (see below). Otherwise, skip the next step and move directly to step 4, using the two FASTA-formatted de-noised files directly.
3. De-noising the pyrosequencing raw data using AmpliconNoise (optional)
If this tutorial is run in a large group, not everybody can do this at once. If you wish, proceed to step 4 and come back to this step later.
Open a web browser and go to http://apps.cbu.uib.no/ampliconnoise. Upload the SFF file and copy / paste the following sequence into the field “Forward primer”: ATTAGATACCCNGGTAG and the following to the field “Samples and barcodes”: “LC_Prefilter, CAGTAGACGT” (enter) and “LC_Final, TGATACGTCT”. See the screenshot below
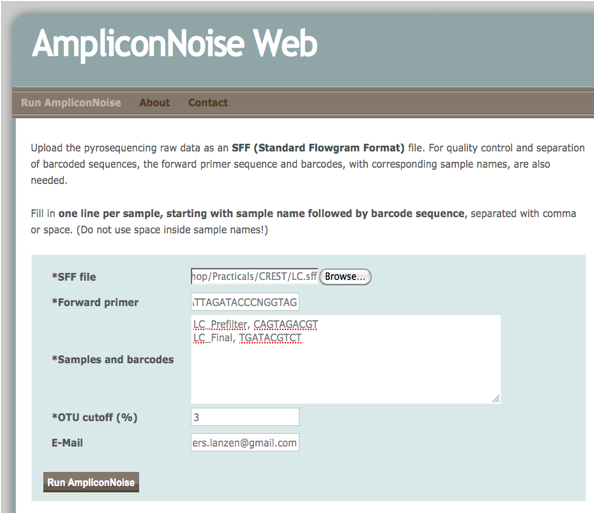
Click Run and wait. If the job takes more than about 10 minutes, then consider having one more coffee, or giving up and moving on to step 4. When the job is finished, download and look at the files AN_Stats.txt and OTU_Diversity_estimates.txt:
-
How many raw reads did you
submit in the SFF file from each dataset?
-
How many reads remain after
filtering and chimera removal?
-
How many unique sequences and
3% OTUs do the remaining reads constitute?
-
What is the Shannon diversity
index?
Download the files OTU_All_Samples.fa and OTU_table.csv for later. Also download
the file Clean_Sequences_w_Abundance.tar.gz
and uncompress it.
This is your de-noised sequences (the same as
those provided already, in case of skipping this step). You can see that each
sequence name ends with an underscore and a number, e.g. “_3”. This indicates
the number of reads representing a unique sequence and can be understood by the
LCAClassifier later on.
4. Aligning files to the reference database (SilvaMod) using Megablast
The first step in the CREST workflow is alignment to a reference database, using Megablast, which is a faster version of BLAST (Basic Local Alignment Search Tool) for nucleotide sequence alignments. We will use the SilvaMod database made from SILVA’s SSURef alignment of full-length SSU rRNA sequences, release 106. Using the resulting alignments (BLAST results), taxonomical classification is then done with the LCAClassifier program. The LCAClassifier only supports results from the NCBI blastall implementation of Megablast, in XML format. It does not work with results from the newer BLAST+ implementation.
Align the sequences in LC_Final_F_Good.fa to SilvaMod using:
megablast -i
LC_Prefilter_F_Good.fa -b 50 -v 50 -m 7 -d
/home/qiime/Desktop/LCAClassifier/parts/flatdb/silvamod/silvamod.fasta -a 2 -o
LC_Prefilter_silvamod.xml
This will take some 5 minutes. Meanwhile, have a look at the the arguments you gave to the megablast command and what they mean:
-i
: the nucleotide sequence input file
-b 50 -v 50 : Report only the 50
best alignments for each sequence
-m 7: Produce output in XML format
-d : the reference database to use*.
-a 2 : Use
two CPU cores.
-o : destination
file of the Megablast output
*Note that the file silvamod.fasta itself is actually missing by default,
but is not needed since MEGABLAST only uses the search index files produced by
the command formatdb from silvamod.fasta.
If you don’t get any error message, the Megablast alignment has probably worked fine. You can also have a look at the resulting file in a text editor, to familiarise yourself with the BLAST XML format, which is the same for Megablast and normal BLAST. However, it is not really intended for being read by human beings and is a bit too structured for us.
Then, align the other FASTA-file:
megablast -i
LC_Final_F_Good.fa -b 50 -v 50 -m 7 -d /home/qiime/Desktop/LCAClassifier/parts/flatdb/silvamod/silvamod.fasta
-a 2 -o LC_Final_silvamod.xml
Again, this takes a few minutes through the QIIME virtual box. Have patience, go for some coffee and relax.
5. Taxonomic classification using LCAClassifier
Classification with the LCAClassifier
program using default parameters is quite easy and carried out using the
command classify. To classify the
Megablast-aligned LC_Prefilter
dataset, simply type:
classify LC_Prefilter_silvamod.xml
This will result in two files. The first, named LC_Prefilter_silvamod_Composition.txt, is a tab-separated text file, listing the number of reads, relative abundance, unique sequences and a Chao-estimate of minimum diversity for each taxon, at different rank levels (domain, phylum, class, order, family and genus). The number of reads classified at each rank level is also summarised. Open this file in a spreadsheet editor (like LibreOffice; make sure to chose only “tab” under “separated by”)
-
What proportion of the original
reads could be classified to at least family level?
-
What is the relative abundance
of Arthrospira (a common alkaliphilic
cyanobacteria)? How many unique sequences are represented in this genus?
-
How many unique sequences were
assigned to the family Rhodobacteraceae and
what is the Chao-estimate for total number of unique sequences?
- How many eukaryotic sequences are there? Can you figure out why, although the primers used target 16S only?
The other file, LC_Prefilter_silvamod_Tree.txt, lists the number of reads assigned in a simple space-delimited tree-format. Here are the first ten lines of the file:
head LC_Prefilter_silvamod_Tree.txt
root: 561
Cellular organisms: 561
Archaea: 27
Euryarchaeota:
27
Methanomicrobia: 9
Methanomicrobiales: 6
Methanocalculus: 6
Methanosarcinales: 3
Methanosaetaceae: 2
Methanosaeta: 2
6. Comparison of composition
between datasets
The LCAClassifier can also classify several Megablast result files at the same time, providing that they were aligned to the same reference database. First delete or rename the old LC_Prefilter_Silvamod_Composition.txt file. Then, try this out:
classify -o *.xml
This results in the same two types of result files for each dataset (LC_Prefilter and LC_Final). However, empty assignments are inserted with zero abundance for each taxon present in at least one dataset, where no assignments to the corresponding taxon was made in a particular dataset. This helps to compare datasets. The option –o instructs the LCAClassifier to also write results in alternative output format (All_Composition.txt).
Open LC_Prefilter_Silvamod_Composition.txt and LC_Final_silvamod_Composition.txt in a spreadsheet editor (e.g. LibreOffice). Copy the last four columns (“Abundance” to “Chao”) of one file into the right of the columns of the other one, so that each taxon is compared for the two datasets side by side.
-
Can you find any taxa
overrepresented in the prefilter sample, with an average abundance > 1% and
less than half in the “final” sample? These are likely to be bulky, filamentous
or non-planktonic organisms.
-
How about a dominant taxon with
almost twice the abundance in the LC_Prefilter
dataset?
-
What is the most diverse genus,
in terms of number of unique de-noised sequences?
-
Have a look at the file All_Composition.txt. Can you figure out
how many sequences that were assigned to the domain Bacteria but could not be
assigned to any particular phylum.
7. Extracting sequences from a taxon
The LCAClassifier can also write output
files that specify the assignment for each individual sequence. Using option -p (or --rdp), the identifier of each sequence
and its predicted taxonomical path is written with suffix “_Assignments.txt”.
For example:
SedimentX_47_5 Cellular organisms;Archaea;Euryarchaeota;
Methanomicrobia;ANME-1;ANME-1a↵
Another useful option is to write a new FASTA file with taxonomic annotations added in the FASTA header. This is done using option -a (or --fasta). By default, only the aligned part of each sequence is written and entries that could not be aligned are omitted. Alternatively, the entire sequences can be written. The file to read in sequences from then must be specified using -i (or --fastain).
We will now produce these files for our representative OTU sequences (Operational Taxonomic Units, basically clusters of similar, unique sequences). Use the file OTU_All_Samples.fa, which contains representative sequences for each OTU in the two datasets:
megablast -i
OTUs_All_Samples.fasta -d /home/qiime/Desktop/LCAClassifier/parts/flatdb/silvamod/silvamod.fasta
-b 50 -v 50 -m7 –a 2 -o OTUs_silvamod.xml
classify -i
OTUs_All_Samples.fasta -a -p OTUs_silvamod.xml
This produces two files with prefix “.fasta” and “.txt” as described above. The first can be used for extracting all sequences from a particular taxon, using the LINUX command grep. For example, to save all archaeal sequences to the file A.fa:
grep Archaea
OTUs_silvamod_Assignments.fasta -A1 > A.fa
-
How many archaea are there?
(Hint: grep -c ‘>’ A.fa)
Some of the sequences are assigned to “Unknown” taxa. This means that the minimum sequence similarity filter of the LCAClassifer has prevented it from being classified to a higher rank. These sequences may be interesting because of their novelty. Have a look at these sequences:
grep Unknown OTUs_silvamod_Assignments.fasta
-A1 > Unknown.fa
The second last sequence (“C56”) is assigned to an unknown order (under Alphaproteobacteria). This means that it is less than 90% similar to the closest reference sequence and thus cannot be assigned to a particular order, but instead to class Alphaproteobacteria. Copy the sequence to NCBI Blast (http://www.ncbi.nlm.nih.gov/blast) and have a look at the closest sequence matches. Note from what type of environment the most similar sequence hit is.
To read an overview of all options available with the LCAClassifier, type:
classify --help
8. Classification using Greengenes
As an alternative to the SilvaMod Taxonomy,
the LCAClassifier can also be used together with the Greengenes Taxonomy.
Similarly to SILVA’s (and “SilvaMod”), this is basically a reference database
consisting of environmental sequences and type strains annotated taxonomically
based on the clustering, or in other words the topology of the distance tree.
The difference from SILVA is that the annotations in Greengenes were made
automatically, using a heuristic algorithm. To read more about the Greengenes
taxonomy, see McDonald et al (2012), 'An improved Greengenes taxonomy with explicit ranks
for ecological and evolutionary analyses of bacteria and archaea', ISME J,
6:610-618.
As the reference
database is different, the first step is to align the sequences to the
Greengenes reference database, which can be found in /home/qiime/Desktop/LCAClassifier/parts/flatdb/greengenes. To align
e.g. the LC_Prefilter sequences:
megablast -i
LC_Final_F_Good.fa -b 50 -v 50 -m 7 -d /home/qiime/Desktop/LCAClassifier/parts/flatdb/greengenes/greengenes.fasta -a 4 -o LC_Final_GG.xml
To classify the sequences based on the Megablast result, you need to tell the classify command to use the Greengenes taxonomy, since SilvaMod is the default.
classify -d greengenes -p LC_Final_GG.xml
Open LC_Prefilter_GG_Composition.txt in a spreadsheet editor.
-
What proportion of reads could
be classified at genus level?
-
Compare it to the community
composition based on the SilvaMod taxonomy. What major differences can you find
between the results based o the two reference databases / taxonomies, on family level?
9. Classification with MEGAN (optional)
In addition to the LCAClassifier, the program MEGAN also supports classification using the CREST scheme. MEGAN has a graphical user interface and many functions for taxonomic and functional classification of metagenomic sequence data. We will revisit these functions in another practical of this course.
By default, MEGAN uses the NCBI taxonomy for assignment. To change this to the SilvaMod taxonomy, select “Use Alternative Taxonomy...” under Edit>Preferences. Then navigate to the directory /home/qiime/Desktop/LCAClassifier/parts/flatdb/silvamod and select the file silvamod.tre. If everything works correctly, the SilvaMod taxonomy should now be enabled and you should see the following output in the Messages window:
Executing: load
treefile='/home/qiime/Desktop/LCAClassifier/parts/flatdb/silvamod/silvamod.tre'
[..]
File name:
/home/qiime/Desktop/LCAClassifier/parts/flatdb/silvamod/silvamod.tre'
Load mapping:
taxId2TaxLevel: 1179077
done: 302383
Load tree:
done: 302379 nodes,
302378 edges
Number of taxa: 302382
Now, import the BLAST results, using File>Import from BLAST. Select one of the Megablast XML files, then go to the sheet LCA Params and change the settings to the following: Min support=1, Min score=155 and Top Percent=2. Also, tick the box “Use Percent Identity Filters”. Then, click Apply!
An interactive taxonomy tree will now appear, listing the number of assignments to different taxa and also symbolising the abundance of a taxon by a circle, whose area is proportional to the number of assignments. Try to explore the tree by collapsing and uncollapsing nodes and by right-clicking a node and selecting Inspect.